
About Me
I am a freelance computer scientist specializing in tools for genomics research. Most of my work is centered on large, collaborative genomics projects such as the Human Pangenome Reference Consortium and Vertebrate Genomes Project.
Current Projects
These are some projects that I am working on now.
Pangenome Reference Graphs
Background
Understanding how and why genotype dictates phenotype is central to the field of genetics. Genotypes are represented as differences between each sample and a single reference genome. The value of this reference is that it provides a common coordinate system that can be used to aggregate data from any individual. But viewing all variation through the lens of a single reference can also lead to bias, for example in cases where all samples under study are much more diverged from the reference than they are to each other. The desire to mitigate reference bias has led to a recent push to use a representative panel of genomes, aka a pangenome, instead of a single reference genome.
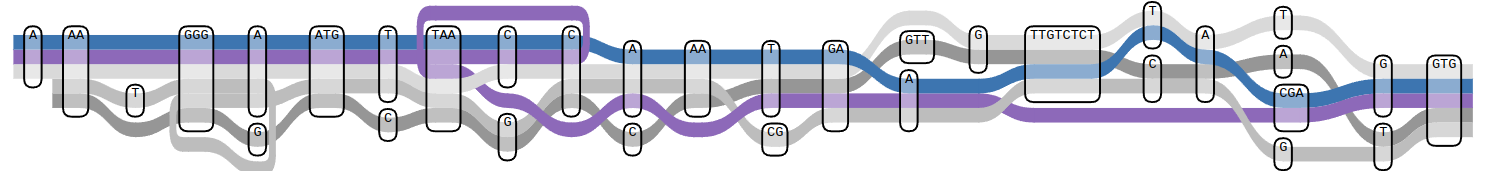
Graph Construction
Pangenomics has undergone a recent paradigm shift, from building graphs from panels of known variants to building graphs directly from genome assemblies. The latter approach further reduces reference bias and can offer better representation of complex sites, but is much more challenging computationally. The Human Pangenome Reference Consortium (HPRC) has as its mission the release of a public pangenome reference comprised of a representative collection of high quality, diploid human genome assemblies. As part of the HPRC I developed a pangenome graph construction pipeline called Minigraph-Cactus (MC) which was used to create reference graphs for the first draft HPRC pangenome and has since become one of, if not the, state of the art pangenome graph construction methods. It is being used in most human pangenome projects, as well as a large number of pangenomes for other species.
Read more about MC here and how it was used in the HPRC here.
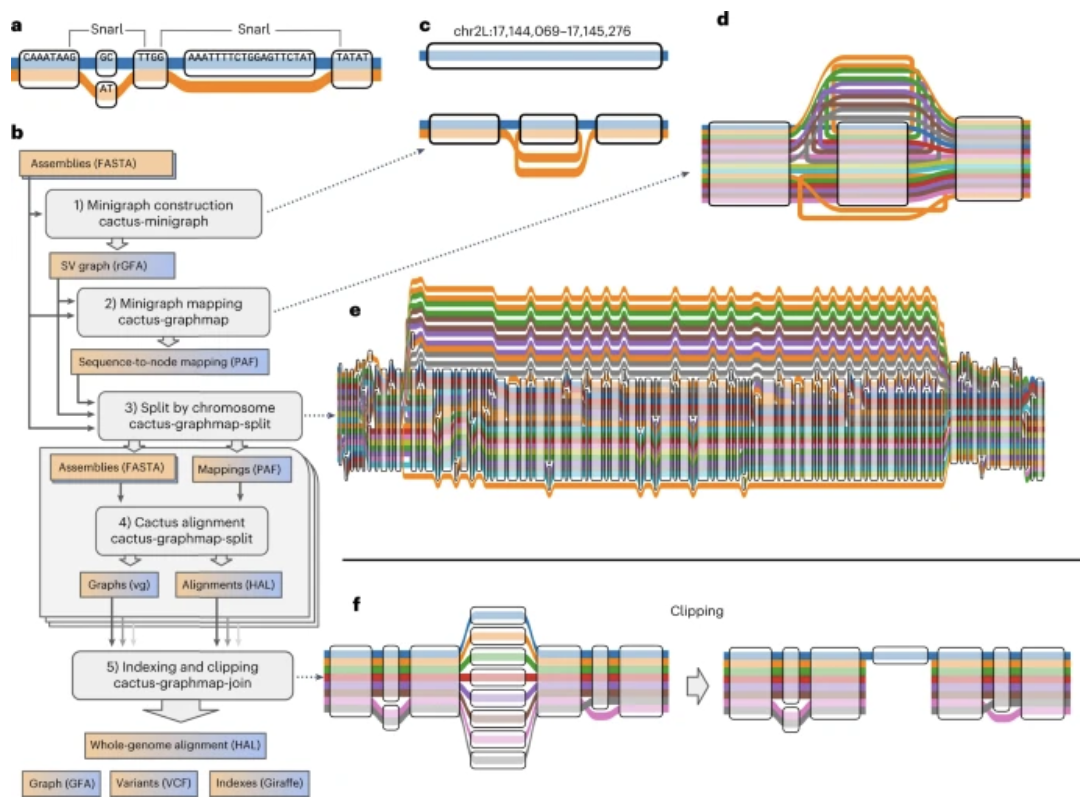
Structural Variants
I have been a principal developer and maintainer of the vg toolkit since shortly after its inception. vg remains among the most widely-used software packages for working with pangenome graphs, and contains tools for graph construction, read mapping and genotyping. One vg tool I created is vg call, which can genotype structural variants in a pangenome graph using support from mapped reads. It remains popular due to its ease of use, accuracy and general applicability.
Read more about SV genotyping with vg here.
Representation of Complex Variants
In theory, pangenome graphs provide an elegant representation of arbitrarily complex variation within a population. In practice, many pangenomics studies end up project their results back to a standard reference genome. They do this to leverage standard formats such as VCF and BAM, as well as the established ecosystem of genomics tools that use them. Projecting back to a single reference leads to many of the pitfalls of reference bias discussed above. To avoid projecting to a reference, I am working on better support for pangenome coordinate systems in general, and their application to common pangenome workflows such as read mapping and genotyping. I case of particular interest is variation nested inside of large SV insertions, which are often “invisible” due limitations of current tools and formats.

Read about an in-progress prototype for nested variation in vg and VCF here, a preprint showing results on HPRC v2 data will be coming soon.
Comparative Genomics
Background
Comparative genomics seeks to use the relationship between species to better understand the relationships and functions of genomes. This is in contrast to pangenomics which primarily concerns variation within species, though genome alignment is a fundamental challenge for both fields. The larger evolutionary distances when aligning between species require different structures, formats and algorithms. Multiple sequence alignment and by extension multiple genome alignment are classical challenges of bioinformatics. They remain active fields as new computing technologies revolutionize the what’s possible with the methods, while the amount of available genome sequences increases exponentially.
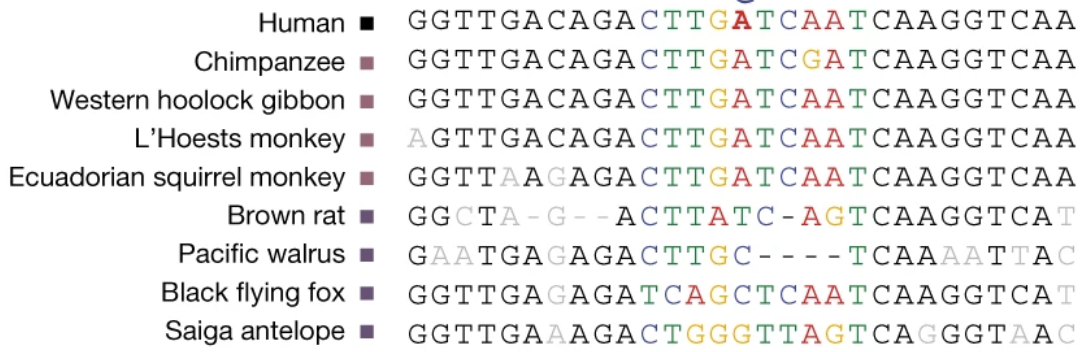
Progressive Cactus Genome Aligner
I am the original and current lead developer of Progressive Cactus, which has remained the state of the art genome aligner for vertebrate and insect genomics studies for several years. Recent genome alignments I have produced with this software have underpinned studies of apes, primates and ants. I am currently preparing the tool to build the first alignment for the Vertebrate Genomes Project (VGP).
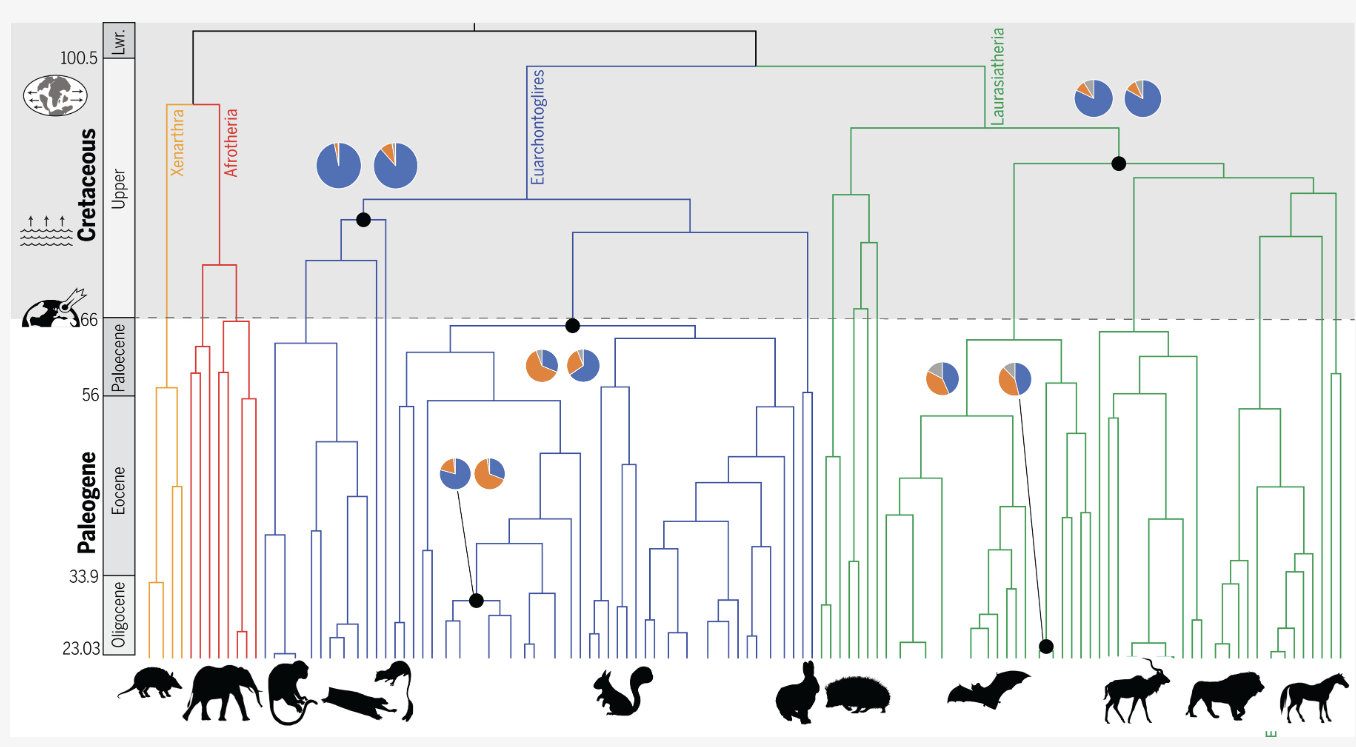
Read about Progressive Cactus here, and the special issue of Science devoted largely to the analysis of the Progressive Cactus Zoonomia alignment here.
Hierarchical Alignment Format (HAL)
Progressive Cactus alignments are made possible by a progressive data format: HAL, which is a row-based file store for genome alignments. It allows for efficient combining and decomposing of huge genome alignments and the associated toolkit provides other basic functionality, including transposition into columnar formats such as MAF which can be used to conservation detection.
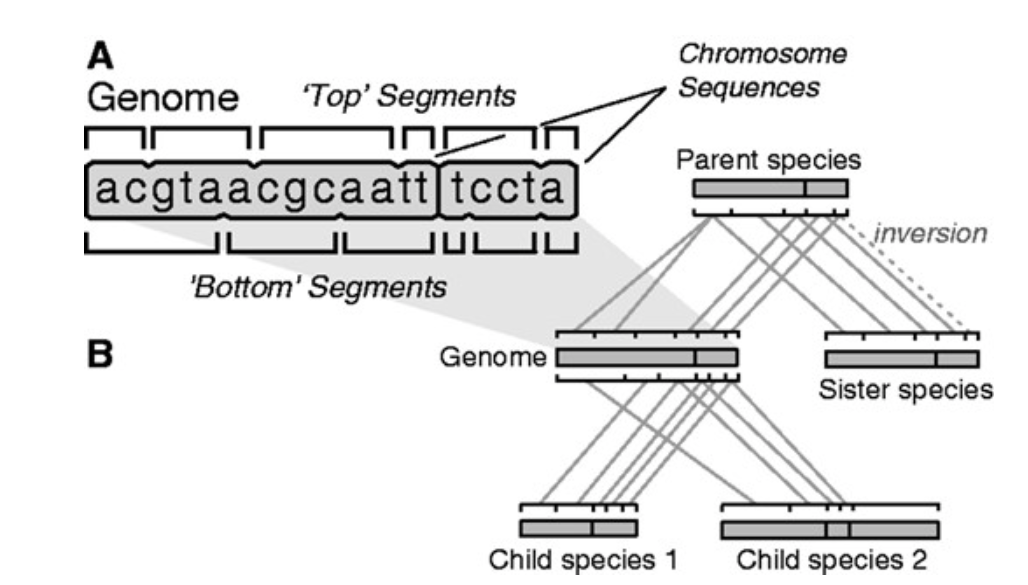
Read about HAL here.
High Performance Computing
The original version of Progressive Cactus was designed to run on the bespoke parasol-based cluster originally developed for the UCSC Genome Browser. Since then, in collaboration with the Toil team and others, I have adapted Cactus to a variety of distributed environments such as AWS EC2, Google Cloud (via terra.bio, Kubernetes and, most recently Slurm. I am currently adapting some of the WDL-based support used for Terra for application to Slurm. I have also worked ot integrate GPU acceleration in the pairwise alignment stage of Progressive Cactus.
Future Projects
These are projects I would like to tackle in the next year (or so) but haven’t quite had the resources to begin in earnest yet
Comparative Pangenomics
Build out an interface between the progressive and pangenome sides of Cactus. The underlying formats allow pangenomes to be used in progressive alignments (early prototypes in this area look promising), but the tooling needs to be modified to better support it. I am particularly interested in applying this for detecting patterns of conservation and incomplete linear sorting (ILS).
HAL 2.0
The HAL format (see above) is based on [HDF5, which is portable and reliable but is also extremely outdated in its lack of multithreading support. I want to change the back end from HDF5 to something based on collections of indexed pairwise alignments. This will be necessary to scale Cactus beyond about 1000 genomes, which will need to happen soon.
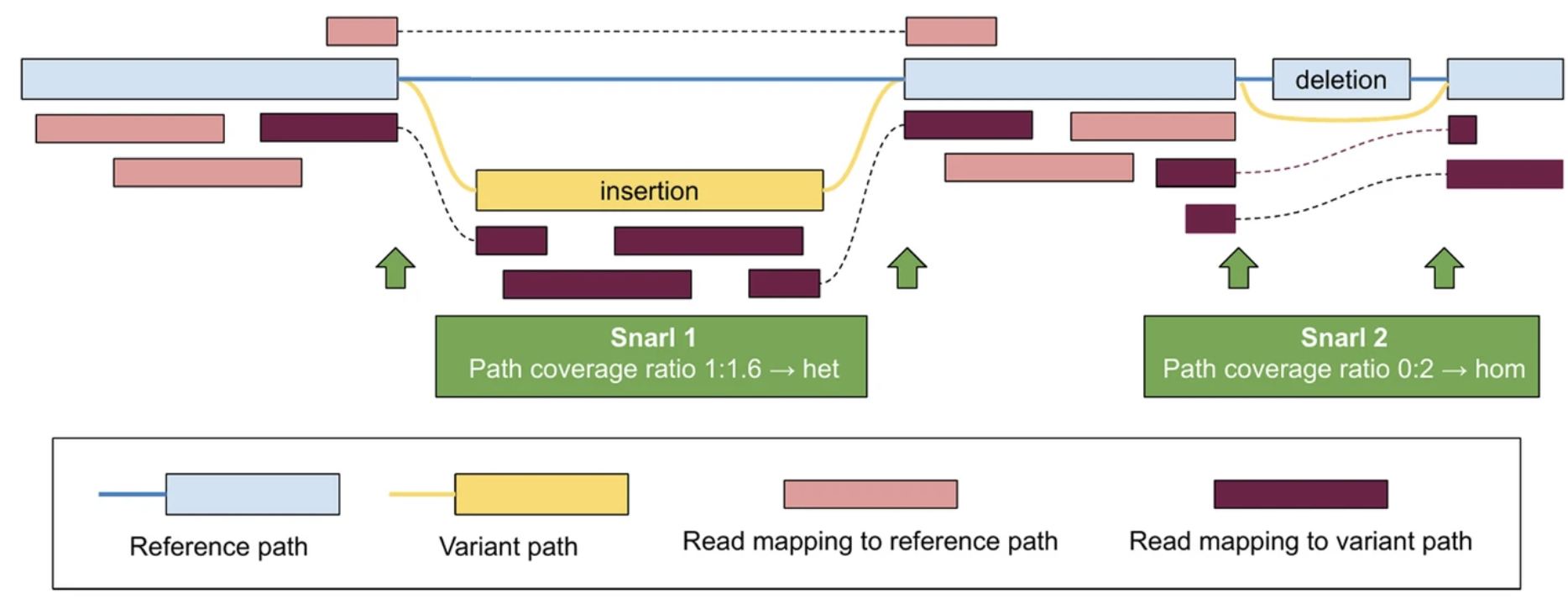
Improved Graph Genotyping
vg call computes a genotype for every site (bubble) in the graph by determining the pair of paths through the site that are most likely given read support. The main drawback of this approach is that read support is determined from pileups on each base and edge in the graph – all longer-range information is discarded. Recent improvements to indexing graph-based mappings should allow the reads themselves (rather than the pileups) to be used for computing genotype likelihoods.
Taffy Tools
The TAF format is an efficient way to store and query alignment columns, and has an interface to efficiently stream windows of alignment data to be consumed by machine learning methods. The tooling to actually use this to detect patterns of conservation, acceleration, incomplete linear sorting, etc. still need to be written.
Haplotype Tagging
Given a mapped read to a pangenome graph, report which haplotypes in the graph best match the read. Provide summary statistics for a set of mapped reads, segmented over the genome. This is a popular feature request, as users want to use the pangenome to determine the ancestry of their own data.